系统树上的某一节点,其分支类群的祖先特征的概率大小,可以通过 MrBayes 计算推断出来。
我们以 MrBayes 软件的示例文件 cynmix.nex 为例,该文件中包含形态学数据和基因序列。
推断祖先特征,其实就是对系统树上某一节点的若干分类群,推断某个形态学特征
(如在序列文件中表示为 0, 1, 2 等)是这一节点类群的共同祖先特征的概率。
本例中,其系统树结构如下图所示:
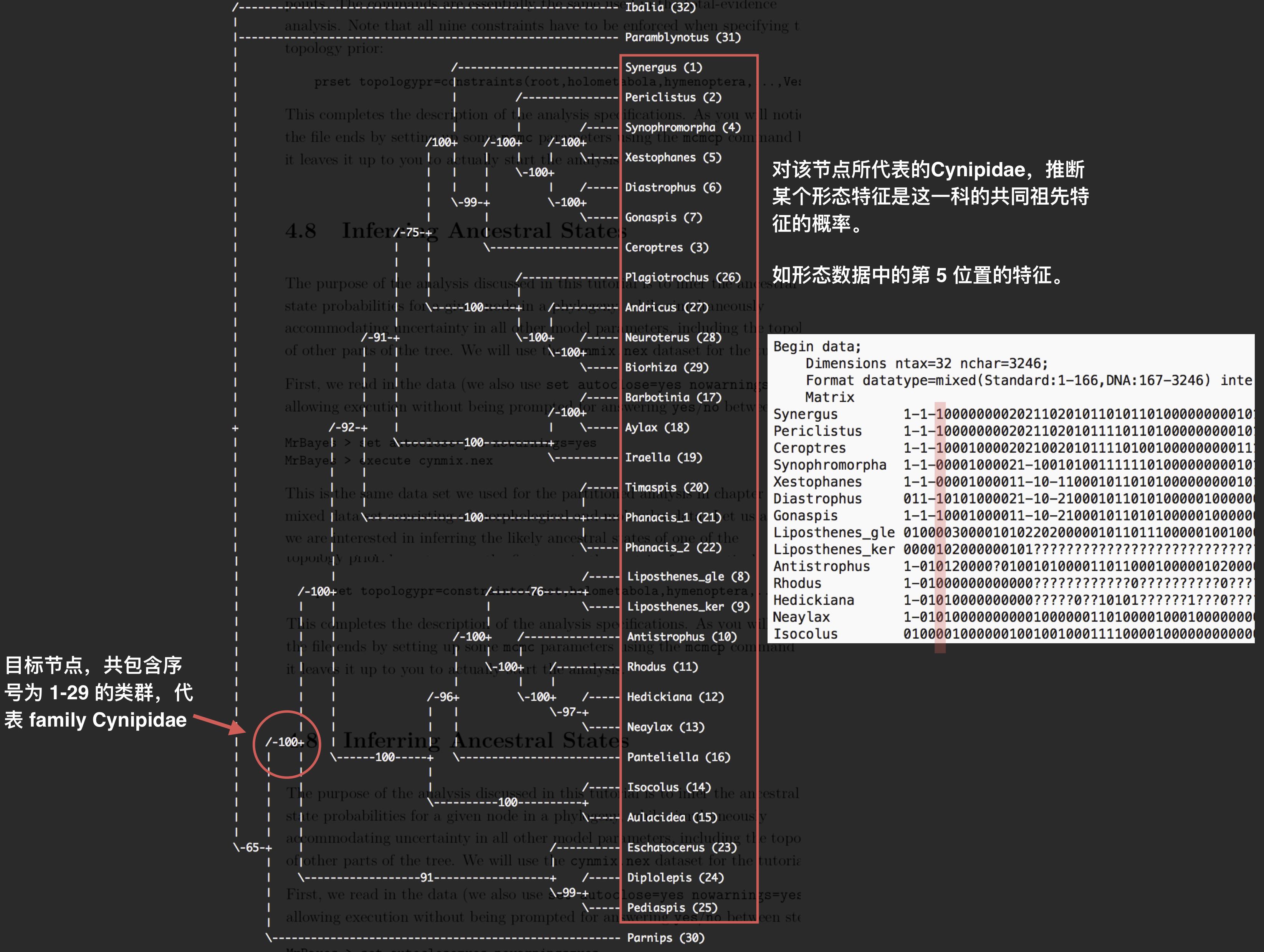
我们即可针对 1-29 的类群所在的节点,推断这些类群的共同祖先特征的概率,本例以第 5 个形态学特征为例,如上图所示, 需要将第 5 个形态学特征分割出来,形成一个单独的分区,因此,必须首先对序列数据重新分区。
MrBayes > execute cynmix.nex
MrBayes > partition ancstates = 6: 5, 1-4 6-166, COI, EF1a, LWRh, 28S
# 源文件的原始分区是 partition ancstates = 5: 1-166, COI, EF1a, LWRh, 28S
# 这里单独挑出第 5 个特征成为单独的一个分区
MrBayes > set partition=ancstates
MrBayes > lset applyto=(2) rates=gamma # 形态学数据使用 gamma 替代率
MrBayes > lset applyto=(3,4,5,6) rates=invgamma nst=6 # 基因数据使用 invgamma 替代率和 GTR 模型
MrBayes > unlink statefreq=(all) revmat=(all) pinvar=(all) shape=(all)
MrBayes > prset ratepr=variable
随后,对 1-29 类群所在的 Cynipidae 科进行树形限制(topological constraint):
MrBayes > constraint cynipidae=1-29
MrBayes > prset topologypr=constraints(cynipidae)
然后,让 MrBayes 报告目标形态特征(第 5 个)是共同祖先特征的概率:
MrBayes > report applyto=(1) ancstates=yes
运行,使用 sump 命令即可在 shell 窗口中查看输出结果:
MrBayes > mcmc ngen=30000
MrBayes > sump
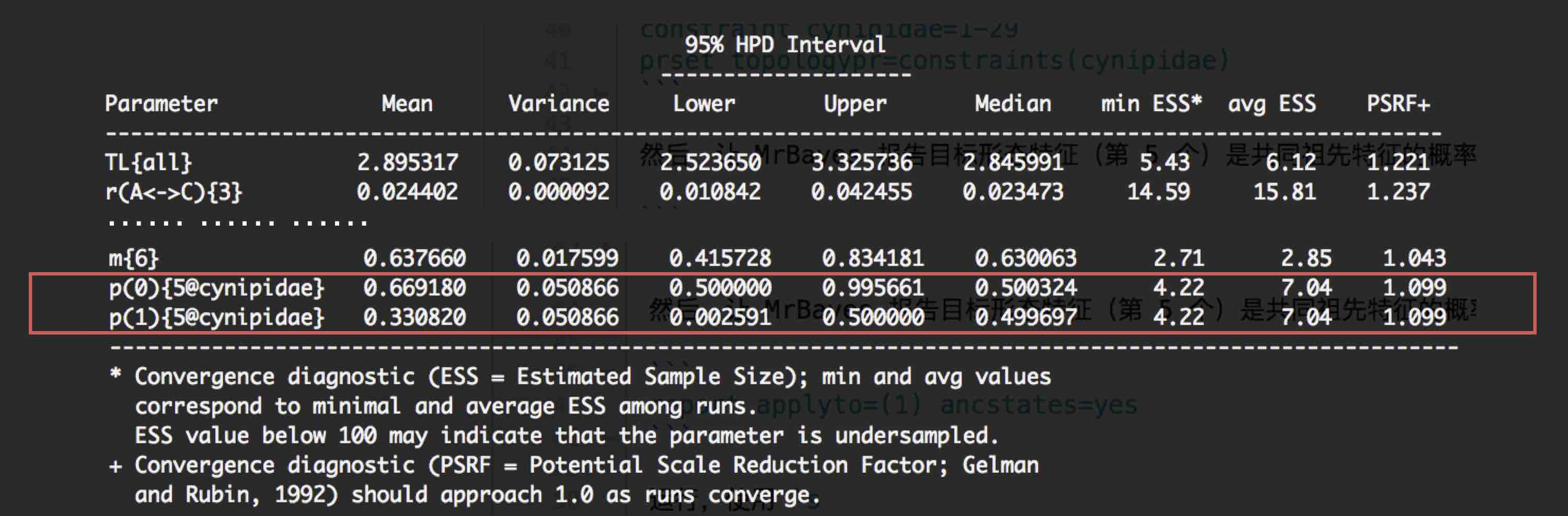
红色框所示的两个参数即表示:节点 cynipidae 的第 5 个形态特征(5@cynipidae)中,
0 所代表的特征与 1 所代表的特征分别是祖先特征的概率(p(0) p(1))。结果发现,
0 所代表的特征是祖先特征的概率比 1 代表的特征更大,因此可以说,第 5 个形态特征中的 0 特征可能是祖先特征。
参考:MrBayes Manual v3.2, p74-76